
检测DNA聚合酶活性的方法及检测试剂盒与流程
本发明涉及生物
技术领域:
,特别是涉及一种检测DNA聚合酶活性的方法及检测试剂盒。
背景技术:
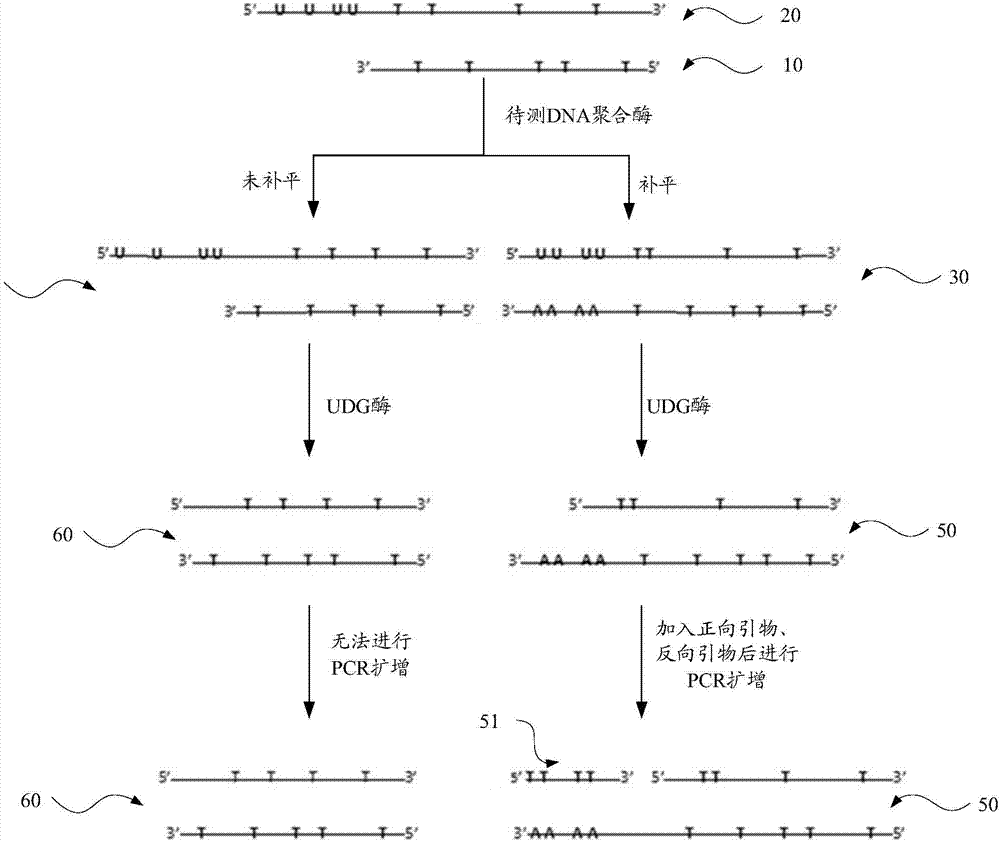
:DNA聚合酶(DNApolymerase)是细胞复制DNA中的重要作用酶,在特定的条件下能够诱导DNA链的复制延长。生物体内较为重要的DNA聚合酶例如有T4DNA聚合酶、Klenow酶(又称Klenow片段,KlenowFragment)和DNA聚合酶I(DNA-polI)等。其中T4DNA聚合酶是由T4噬菌体聚合酶I基因编码,具有5’-3’聚合酶活性以及3’-5’外切酶活性。T4DNA聚合酶是基因工程技术中一种重要的工具酶,在高通量测序(nextgenerationsequencing)文库构建的过程具有重要的作用。传统的检测DNA聚合酶活性的方法一般采用放射性同位素法,将DNA聚合酶催化的32P标记的dNTP掺入到小牛胸腺DNA中。经过TCA沉淀、whatman滤纸过滤,通过测定酸不溶性物质中放射性的含量,从而计算聚合酶活性。这种方法易产生放射性污染、操作繁琐,难以实现高通量的检测。技术实现要素:基于此,有必要提供一种非放射性、操作简便的检测DNA聚合酶活性的方法及检测试剂盒。一种检测DNA聚合酶活性的方法,包括以下步骤:提供待补平模板,所述待补平模板包括第一模板链和第二模板链,所述第一模板链包括第一DNA片段,所述第二模板链包括第二DNA片段以及与所述第二DNA片段5’端相连接的凸出片段,所述第二DNA片段与所述第一DNA片段碱基互补,所述凸出片段中含有U碱基;在dNTP存在的条件下,将待测DNA聚合酶与足量的所述待补平模板混合,使得所述第一DNA片段的3’端延伸形成补平片段,得到补平模板,所述补平片段与所述凸出片段碱基互补;将所述补平模板与正向引物、反向引物、探针、dNTP以及足量的UDG酶混合进行实时荧光定量PCR扩增反应,其中所述正向引物针对所述凸出片段设计,所述反向引物针对所述第一DNA片段的5’端设计,所述探针用于检测所述第一DNA片段;以及获取所述实时荧光定量PCR扩增反应的CT值,将所述CT值带入由DNA聚合酶标准品建立的标准曲线中,得到所述待测DNA聚合酶的酶活性。在一个实施方式中,所述由DNA聚合酶标准品建立的标准曲线通过如下操作得到:将DNA聚合酶标准品进行梯度稀释,获得梯度酶活性的所述DNA聚合酶标准品;在dNTP存在的条件下,分别将每一个酶活性的所述DNA聚合酶标准品与足量的所述待补平模板混合,得到多个补平模板,所述多个补平模板分别为每一个酶活性的所述DNA聚合酶标准品对应的补平模板;分别将所述多个补平模板与正向引物、反向引物、探针、dNTP以及足量的UDG酶混合进行实时荧光定量PCR扩增反应;以及获取每一个酶活性的所述DNA聚合酶标准品对应的实时荧光定量PCR扩增反应的CT值,根据所述CT值与酶活性的对应关系建立所述标准曲线。在一个实施方式中,所述第一模板链的碱基序列如SEQIDNo.1所示,所述第二模板链的碱基序列如SEQIDNo.2所示。在一个实施方式中,所述正向引物的碱基序列如SEQIDNo.3所示,所述反向引物的碱基序列如SEQIDNo.4所示。在一个实施方式中,所述探针的碱基序列如SEQIDNo.5所示。在一个实施方式中,所述将待测DNA聚合酶与足量的所述待补平模板混合的操作中,混合的温度为25℃~40℃,混合的时间为0.5h~1.5h。在一个实施方式中,所述将所述补平模板、正向引物、反向引物、探针、dNTP以及足量的UDG酶混合进行实时荧光定量PCR扩增反应的操作中,所述荧光定量PCR扩增反应的热循环条件为:45℃~55℃、20min~40min,1~3个循环;93℃~97℃、5min~15min,1~3个循环;以及90℃~100℃、15s~25s,50℃~60℃、15s~25s,70℃~75℃、25s~35s,35~45个循环。在一个实施方式中,所述DNA聚合酶选自T4DNA聚合酶、Klenow酶和DNA聚合酶I中的至少一种。一种检测DNA聚合酶活性的检测试剂盒,包括待补平模板、正向引物、反向引物和探针;所述待补平模板包括第一模板链和第二模板链,所述第一模板链包括第一DNA片段,所述第二模板链包括第二DNA片段以及与所述第二DNA片段5’端相连接的凸出片段,所述第二DNA片段与所述第一DNA片段碱基互补,所述凸出片段中含有U碱基;所述正向引物针对所述凸出片段设计;所述反向引物针对所述第一DNA片段的5’端设计;所述探针针对所述第一DNA片段中的部分碱基设计。在一个实施方式中,第一模板链的碱基序列如SEQIDNo.1所示,所述第二模板链的碱基序列如SEQIDNo.2所示,所述正向引物的碱基序列如SEQIDNo.3所示,所述反向引物的碱基序列如SEQIDNo.4所示,所述探针的碱基序列如SEQIDNo.5所示。上述检测DNA聚合酶活性的方法,在dNTP存在的条件下,将待测DNA聚合酶与足量的待补平模板混合后,通过待测DNA聚合酶诱导待补平模板中的第一DNA片段的3’端延伸形成与凸出片段的碱基互补的补平片段,从而得到补平模板。将补平模板、正向引物、反向引物、探针、dNTP以及足量的UDG酶混合进行实时荧光定量PCR扩增反应。补平模板中的U碱基被足量的UDG酶消化去除,得到适于PCR扩增的模板,正向引物针对凸出片段设计,反向引物针对第一DNA片段的5’端设计,即正向引物能够结合补平片段,诱导第一模板链扩增。而未经补平的模板由于没有形成补平片段,经消化酶去除U碱基后则无法与正向引物配对,即不能进行PCR扩增反应。经研究发现,不同酶活性的DNA聚合酶和实时荧光定量PCR扩增反应的CT值(荧光信号到达设定域值时所经历的循环数)存在稳定的相关性,在足量的待补平模板的条件下,DNA聚合酶的酶活性越高,诱导补平后获得的补平模板的量越多,经消化酶酶切后的获得的适于PCR扩增的模板的量也越多,PCR扩增反应的CT值越小。将待测DNA聚合酶的CT值带入由DNA聚合酶标准品建立的标准曲线中,运算即可获得待测DNA聚合酶的酶活性。上述检测DNA聚合酶活性的方法不需要放射性检测,操作简便,实验结果表明该方法重现性好、灵敏度高,能够实现高通量的检测。附图说明图1为一实施方式的检测DNA聚合酶活性的方法的流程图;图2为一实施方式的检测DNA聚合酶活性的方法的原理示意图;图3为不同稀释倍数的T4DNA聚合酶标准品PCR扩增后的扩增曲线;图4为以T4DNA聚合酶标准品的酶活性作为横坐标,PCR扩增反应对应的CT值作为纵坐标绘制的标准曲线;图5为以另一种T4DNA聚合酶标准品的酶活性作为横坐标,PCR扩增反应对应的CT值作为纵坐标绘制的标准曲线;图6为以自制的DNA聚合酶的酶活性作为横坐标,PCR扩增反应对应的CT值作为纵坐标绘制的标准曲线。具体实施方式为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合具体实施例及附图对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施的限制。未特别说明,序列表中的碱基序列均为从5’端到3’端的顺序。请参阅图1,一实施方式的检测DNA聚合酶活性的方法包括以下步骤S110~S140。S110、提供待补平模板,该待补平模板包括第一模板链和第二模板链,第一模板链包括第一DNA片段,第二模板链包括第二DNA片段以及与第二DNA片段5’端相连接的凸出片段,第二DNA片段与第一DNA片段碱基互补,凸出片段中含有U碱基。当第一模板链和第二模板链形成双链后,由于第二DNA片段与第一DNA片段碱基互补,第二模板链中的凸出片段形成凸出末端,而第一模板链中的3’端凹陷。S120、在dNTP存在的条件下,将待测DNA聚合酶与足量的S110中的待补平模板混合,使得第一DNA片段的3’端延伸形成补平片段,得到补平模板,补平片段与凸出片段碱基互补。DNA聚合酶是细胞复制DNA中的重要作用酶,在特定的条件下能够诱导DNA链的复制延长,DNA聚合酶一般以DNA为复制模板,将DNA由5"端向3"端延伸。一实施方式检测的DNA聚合酶选自T4DNA聚合酶、Klenow酶和DNA聚合酶I中的至少一种。具体在本实施方式中,检测的DNA聚合酶为T4DNA聚合酶。具体地,待补平模板的量以及加入的dNTP(脱氧核糖核苷三磷酸,包括dATP、dGTP、dTTP、dCTP)为足量的,即不会因为待补平模板或dNTP被消耗掉而导致反应的终止,待补平模板或dNTP可能是过量的也有可能是刚刚好。请参阅图2,检测DNA聚合酶活性的方法的原理示意图,待补平模板包括第一模板链10和第二模板链20,第一模板链10包括第一DNA片段,第二模板链20包括第二DNA片段以及与第二DNA片段5’端相连接的凸出片段,凸出片段中含有U碱基。第一模板链10和第二模板链20形成双链后,第一DNA片段从5’端到3’端的碱基与第二DNA片段从3’端到5’端的碱基互补。由于第二模板链20含有凸出片段,形成双链后第二模板链20的5’端凸出而第一模板链10的3’端凹陷。在dNTP存在的条件下,待测DNA聚合酶与足量的待补平模板混合,通过待测DNA聚合酶诱导待补平模板中的第一DNA片段延伸形成与凸出片段碱基互补的补平片段。从而得到补平模板30,补平模板30为末端平滑的双链模板。由于待补平模板的量为足量的,待测DNA聚合酶量相对不足,反应后体系中还存在未补平的双链40。在一个实施方式中,第一模板链从5’端到3’端的碱基序列如SEQIDNo.1所示,而从3’端到5’端的碱基序列为:TAGCACCGTTCGACATGAGATAGGCAAGTCGCCGTCGGAAACCGATGCCATCGAGTCGACGAGATAGATAATAAGCTAGAATAATAATATAAA。第二模板链的序列如SEQIDNo.2所示,在第二模板链中第二DNA片段的从5’端到3’端的碱基序列为:ATCGTGGCAAGCTGTACTCTATCCGTTCAGCGGCAGCCTTTGGCTACGGTAGCTCAGCTGCTCTATCTATTATTCGATCTTATTATTATATTT。与第二DNA片段5’端相连接的凸出片段的序列为:CAUAUAGCAUAUCGAUCGUAGCTG。如SEQIDNo.1所示的第一模板链以及如SEQIDNo.2所示的第二模板链,第一DNA片段从5’端到3’端的碱基与第二DNA片段从3’端到5’端的的碱基互补。通过待测DNA聚合酶诱导待补平模板中的第一DNA片段的3’端延伸形成补平片段,该补平片段从3’端到5’端的序列为GTATATCGTATAGCTAGCATCGAC。补平片段从3’端到5’端与凸出片段从5’端到3’端碱基互补。设计的第二模板链和第二模板链长度适宜,凸出片段的碱基数量合适,便于根据该模板链设计适宜的扩增引物和探针。实验结果表明,序列如SEQIDNo.1所示的第二模板链和序列如SEQIDNo.2所示的第二模板链组成的待补平模板能够特异性的结合DNA聚合酶,从而在待测DNA聚合酶的诱导下得到补平模板。在一个实施方式中,在将待测DNA聚合酶与足量的待补平模板混合的操作之前,还包括将待测DNA聚合酶的样品经过SDS-PAGE跑胶,根据SDS-PAGE测定的蛋白浓度,初步确定样品中DNA聚合酶的酶活性,从而确定待补平模板的量,提高检测的准确性。在一个实施方式中,检测的DNA聚合酶为T4DNA聚合酶,T4DNA聚合酶是由T4噬菌体聚合酶I基因编码,具有5’-3’聚合酶活性以及3’-5’外切酶活性。T4DNA聚合酶是基因工程技术中一种重要的工具酶,在高通量测序(nextgenerationsequencing)文库构建的过程具有重要的作用。本实施方式利用T4DNA聚合酶具有5’-3’聚合酶活性诱导待补平模板中的第一DNA片段的3’端延伸形成与凸出片段的碱基互补的补平片段,从而得到补平模板。补平模板经消化酶酶切去除U碱基后与引物配对,从而进行PCR扩增反应,根据PCR扩增反应的CT值检测DNA聚合酶的活性,检测重现性好,即使0.3×10-6U/μL较低的酶活性依然能够检测出来,灵敏度高。在一个实施方式中,将待测DNA聚合酶与足量的待补平模板混合的操作中,混合温度为25℃~40℃,反应时间为0.5h~1.5h,例如37℃下反应1h。S130、将S120中得到的补平模板与正向引物、反向引物、探针、dNTP以及足量的UDG酶混合进行实时荧光定量PCR扩增反应,其中正向引物针对凸出片段设计,反向引物针对第一DNA片段的5’端设计,探针用于检测第一DNA片段。具体地,UDG酶(尿嘧啶-DNA糖基化酶)能够消化双链模板中的U碱基,切去部分凸出片段。请继续参阅图2,补平模板30经UDG酶去除U碱基后,得到扩增模板50。加入正向引物和反向引物后,扩增模板50中由第一DNA片段的3’端延伸形成补平片段特异性的与正向引物51互补配对,从而进行PCR扩增反应。由于正向引物针对凸出片段设计,反向引物针对第一DNA片段的5’端设计。正向引物能够与第一DNA片段的3’端延伸形成补平片段互补,反向引物针对第一DNA片段的5’端设计,反向引物能够与第一DNA片段复制后形成的互补链的3’端互补,使得含有第一DNA片段的模板链实现指数扩增。而未补平的双链40经消化酶酶切去除双链模板中的U碱基后,得到一般的DNA双链60,该DNA双链60中不含有补平片段,从而无法与正向引物51配对,即不能进行PCR扩增反应。在足量的待补平模板的条件下,DNA聚合酶的酶活性越高,诱导补平后获得的补平模板30的量越多,酶切后的获得的扩增模板50的量也越多,PCR扩增反应的荧光信号到达设定域值时所经历的循环数相应的越少,即CT值越小。相反,DNA聚合酶的酶活性越低,诱导补平后获得的补平模板30的量越少,酶切后的获得的扩增模板50的量也越少,PCR扩增反应的荧光信号到达设定域值时所经历的循环数相应的越多,即CT值越大。将待测DNA聚合酶与相应的DNA聚合酶标准品所经历的循环数比较运算即可获得待测DNA聚合酶的酶活性。具体地,第一模板链的碱基序列如SEQIDNo.1所示,第二模板链的碱基序列如SEQIDNo.2所示。经足量的消化酶酶切去除双链模板中的U碱基后,SEQIDNo.2中的CAUAUAGCAUAUCGAUCGU片段被去除。在一个实施方式中,正向引物的碱基序列与凸出片段中的部分或全部的碱基序列相同,反向引物的碱基序列与第一DNA片段5’端的部分碱基序列相同。正向引物针对凸出片段设计,从而与第一DNA片段的3’端延伸形成补平片段互补,特异性诱导扩增模板中含有第一DNA片段的这一条DNA单链复制扩增。反向引物针对第一DNA片段的5’端设计,与第一DNA片段中的部分碱基序列相同。在前期的扩增反应中,能够与少量的第二DNA片段中部分碱基序列互补,诱导扩增模板中含有第二DNA片段的这一条DNA单链复制扩增。之后,反向引物与第一DNA片段复制后形成的互补链的3’端互补,即诱导第一DNA片段的复制后形成的互补链的扩增。正向引物与反向引物配合,使得含有第一DNA片段的模板链实现指数扩增。具体地,正向引物和反向引物的摩尔比为0.5~2:1,例如1:1。具体地,正向引物从5’端到3’端的碱基序列如SEQIDNo.3所示,反向引物从5’端到3’端的碱基序列如SEQIDNo.4所示。正向引物和反向引物碱基数合理,快速诱导扩增模板进行扩增。具体地,探针用于检测第一DNA片段,探针的两端分别结合有荧光报告基团和荧光淬灭基团。探针的碱基序列与第一DNA片段中的部分碱基序列相同,探针能够捕获扩增后形成的扩增产物,根据荧光强度实时监控PCR反应的进程。在一个实施方式中,探针从5’端到3’端的碱基序列如SEQIDNo.5所示。探针碱基数合理,能够特异性的捕获扩增后形成的扩增产物。具体地,荧光报告基团结合在探针的5’端,荧光淬灭基团结合在探针的3’端。荧光报告基团例如为FAM(6-羧基荧光素),荧光淬灭基团例如为BHQ1。具体地,正向引物、反向引物以及探针的摩尔比为0.5~2:0.5~2:1,例如1:1:1。在一个实施方式中,将补平模板与正向引物、反向引物、探针、dNTP以及足量的UDG酶混合进行实时荧光定量PCR扩增反应的操作中,除加入正向引物、反向引物、探针、dNTP以及足量的UDG酶外,还包括加入耐高温DNA聚合酶。耐高温DNA聚合酶例如为HotsartTaq酶,使得DNA扩增过程中保持高保真度。在一个实施方式中,将补平模板、正向引物、反向引物、探针、dNTP以及足量的UDG酶混合进行实时荧光定量PCR扩增反应的操作中,荧光定量PCR扩增反应的热循环条件为:45℃~55℃、20min~40min,1~3个循环,93℃~97℃、5min~15min,1~3个循环;以及90℃~100℃、15s~25s,50℃~60℃、15s~25s,70℃~75℃、25s~35s,35~45个循环。进一步地,荧光定量PCR扩增反应的热循环条件为:50℃、30min,1个循环;95℃、10min,1个循环;94℃、20s,55℃、20s,72℃、30s,40个循环。具体地,50℃、30min,1个循环使得UDG酶在适宜的条件下去除补平模板中U碱基,得到扩增模板。95℃、10min,1个循环,使得扩增模板中地双链打开。94℃、20s,55℃、20s,72℃、30s,40个循环,PCR循环扩增条件,在PCR循环扩增中能够收集到荧光。S140、获取实时荧光定量PCR扩增反应的CT值,将CT值带入由DNA聚合酶标准品建立的标准曲线中,得到待测DNA聚合酶的酶活性。在足量的待补平模板的条件下,DNA聚合酶的酶活性越高,诱导补平后获得的补平模板的量越多,UDG酶切后的获得的扩增模板的量也越多,在PCR扩增反应中,PCR扩增反应的荧光信号到达设定域值时所经历的循环数相应的越少,即PCR扩增反应的CT值越小。因此通过将待测DNA聚合酶与相应的DNA聚合酶标准品所经历的循环数比较运算即可获得待测DNA聚合酶的酶活性。具体地,PCR扩增反应的CT值(Cycle)为PCR扩增反应中荧光信号开始由本底进入指数增长阶段所对应的循环数。研究结果表明,不同酶活性的DNA聚合酶和CT值的对数具有良好的线性关系,因此通过将待测DNA聚合酶与相应的DNA聚合酶标准品经相同的补平条件和酶切条件后获得扩增模板,比较两者扩增模板PCR扩增反应中所经历的循环数即可获得待测DNA聚合酶的酶活性。在一个实施方式中,由DNA聚合酶标准品建立的标准曲线通过如下操作得到:将DNA聚合酶标准品进行梯度稀释,获得梯度酶活性的DNA聚合酶标准品。具体地,进行梯度稀释所用的稀释液例如为TE缓冲液。在dNTP存在的条件下,分别将每一个酶活性的DNA聚合酶标准品与足量的待补平模板混合,得到多个补平模板,其中多个补平模板分别为每一个酶活性的DNA聚合酶标准品对应的补平模板。分别将多个补平模板与正向引物、反向引物、探针、dNTP以及足量的UDG酶混合进行实时荧光定量PCR扩增反应。获取每一个酶活性的DNA聚合酶标准品对应的实时荧光定量PCR扩增反应的CT值,根据CT值与酶活性的对应关系建立标准曲线。检测时,将待测DNA聚合酶在对应的在PCR扩增反应中获得的CT值带入标准曲线中运算即可获得待测DNA聚合酶的活性。上述检测DNA聚合酶活性的方法为非放射性的检测,操作简便。待测DNA聚合酶诱导待补平模板中的第一DNA片段延伸形成与凸出片段的碱基互补补平片段,从而将待补平模板补平形成补平模板。将补平模板、正向引物、反向引物、探针、dNTP以及足量的UDG酶混合进行实时荧光定量PCR扩增反应。补平模板中的U碱基被足量的UDG酶消化去除,得到适于PCR扩增的模板,正向引物针对凸出片段设计,反向引物针对第一DNA片段的5’端设计,即正向引物能够结合补平片段,诱导第一模板链扩增。而未经补平的模板由于没有形成补平片段,经消化酶去除U碱基后则无法与正向引物配对,即不能进行PCR扩增反应。根据不同酶活性的DNA聚合酶和PCR扩增反应的CT值与相应的DNA聚合酶标准品的CT值比较运算即可获得待测DNA聚合酶的酶活性。实验结果表明该方法重现性好、灵敏度高,能够实现高通量的检测。一实施方式的检测DNA聚合酶活性的检测试剂盒包括待补平模板、正向引物、反向引物和探针。其中,待补平模板包括第一模板链和第二模板链,第一模板链包括第一DNA片段,第二模板链包括第二DNA片段以及与所述第二DNA片段5’端相连接的凸出片段,第二DNA片段与第一DNA片段碱基互补,凸出片段中含有U碱基。正向引物针对凸出片段设计,具体地,正向引物的碱基序列与凸出片段中的部分或全部的碱基序列相同。反向引物针对第一DNA片段的5’端设计,具体地,反向引物的碱基序列与第一DNA片段5’端的部分碱基序列相同。探针针对第一DNA片段中的部分碱基设计。PCR扩增时,探针用于检测第一DNA片段。探针的碱基序列与第一DNA片段中的部分碱基序列相同。在一个实施方式中,第一模板链从5’端到3’端的序列如SEQIDNo.1所示,第二模板链从5’端到3’端的序列如SEQIDNo.2所示。设计的第一模板链和第二模板链长度适宜,凸出片段的碱基数量合适,便于根据该模板链设计适宜的扩增引物和探针。实验结果表明,序列如SEQIDNo.1所示的第一模板链和序列如SEQIDNo.2所示的第二模板链组成的待补平模板能够特异性的结合DNA聚合酶,从而在待测DNA聚合酶的诱导下得到补平模板。具体地,正向引物从5’端到3’端的碱基序列如SEQIDNo.3所示,反向引物从5’端到3’端的碱基序列如SEQIDNo.4所示。具体地,探针从5’端到3’端的碱基序列如SEQIDNo.5所示。具体地,荧光报告基团结合在探针的5’端,荧光淬灭基团结合在探针的3’端。荧光报告基团例如为FAM(6-羧基荧光素),荧光淬灭基团例如为BHQ1。具体地,使用时,正向引物、反向引物以及探针的的摩尔比为0.5~2:0.5~2:1,例如1:1:1。上述检测试剂盒可用于检测T4DNA聚合酶、Klenow酶和DNA聚合酶I中的至少一种DNA聚合酶的酶活性。上述检测试剂盒用于检测DNA聚合酶活性时,不需要放射性的试剂,操作简便、快速,检测的重现性好、灵敏度高。以下为具体实施例部分。实施例中采用试剂和仪器如非特别说明,均为本领域常规选择。实施例中未注明具体条件的实验方法,通常按照常规条件,例如文献、书本中所述的条件或者试剂盒生产厂家推荐的方法实现。实施例1由T4DNA聚合酶标准品建立的标准曲线1.1、提供包括第一模板链和第二模板链的待补平模板,第一模板链的碱基序列如SEQIDNo.1所示,第二模板链的碱基序列如SEQIDNo.2所示。取10μL的T4DNA聚合酶标准品(3U/μL)加入90μL的1×TE缓冲液中,进行连续6个10倍梯度稀释,形成6个标准品梯度。分别按下表1配置补平反应体系,补平反应条件为:37℃,1h,补平后得到补平模板。表1:补平反应体系试剂体积10×T4DNA聚合酶缓冲液5μLdNTP(10mmol/L)0.2μL待补平模板(0.025μmol/L)10μL梯度稀释T4DNA聚合酶5μLddH2O补充至总体积为50μL其中,T4DNA聚合酶标准品为商品化T4DNA聚合酶(NEB-T4DNAPolymerase)。10×的T4DNA聚合酶缓冲液的配方如下:NaCl500mmol/L、Tris-HCl100mmol/L、MgCl2100mmol/L、DTT10mmol/L,25℃下pH为7.9。1.2、取10μL1.1中补平后的体系按下表2配置PCR反应体系,其中,正向引物从5’端到3’端的碱基序列如SEQIDNo.3所示。反向引物从5’端到3’端的碱基序列如SEQIDNo.4所示。探针从5’端到3’端的碱基序列如SEQIDNo.5所示,探针的5’端结合有荧光报告基团FAM,3’端结合有荧光淬灭基团BHQ1。表2:PCR反应体系1.3、将按表2配置PCR反应体系,每个梯度稀释的样品至少两份样品,置于荧光定量PCR扩增仪中,按下表3设置PCR扩增条件,并实时检测荧光亮度,获得扩增曲线。表3:PCR扩增条件不同稀释倍数的T4DNA聚合酶标准品的PCR后的扩增曲线如图3所示,对应的CT值如下表4。表4:不同稀释倍数的T4DNA聚合酶标准品的CT值从结果可以看出,在足量的待补平模板的条件下,T4DNA聚合酶的酶活性越高,诱导补平后获得的补平模板的量越多,酶切后的获得的扩增模板的量也越多,PCR扩增反应的对应的CT值越小。相反,T4DNA聚合酶的酶活性越低,诱导补平后获得的补平模板的量越少,酶切后的获得的扩增模板的量也越少,PCR扩增反应的对应的CT值越大。以T4DNA聚合酶标准品的酶活性为横坐标,PCR扩增反应对应的CT值为纵坐标用GraphPadPrism5作图得到图4,T4DNA聚合酶标准品的酶活性与CT值呈对数型线性关系,得到曲线方程为y=-0.876ln(x)+13.235,R2=0.9982。实施例2检测未知T4DNA聚合酶的酶活性将待检样品SDS-PAGE跑胶,根据蛋白浓度初步标定待检样品中T4DNA聚合酶酶活。取10μL待检样品进行100倍稀释,平行稀释8组。根据实施例1中表1配制补平体系,按照表2的PCR反应体系配置PCR反应体系,并根据表3中的PCR扩增条件进行PCR扩增,获取8组样品PCR扩增对应的CT值,结果如表5所示。表5:T4DNA聚合酶样品的CT值样品标号1#2#3#4#5#6#7#8#CT值16.6716.6816.6116.6516.6716.6216.5816.6将样品对应的CT值分别带入图4的曲线方程中,推算待检样品中T4DNA聚合酶的酶活,结果如下表6。表6:样品中T4DNA聚合酶的酶活性通过多次重复实验,并计算曲线的相关系数和待检样品的酶活,我们发现,相关系数和待检样品的酶活多次实验间保持稳定,重现性好。实施例3检测不同来源的T4DNA聚合酶的酶活性为了进一步验证检测方法的可行性,选择两种商品化T4DNA聚合酶进行检测,商品化T4DNA聚合酶1为NEB-T4DNAPolymerase,商品化T4DNA聚合酶2为Enzymatics-T4DNAPolymerase。另外还检测自行表达纯化T4DNA聚合酶的酶活性,自制T4DNA聚合酶克隆自T4噬菌体DNA聚合酶I基因,克隆至pET28a表达载体并转化大肠杆菌BL21进行表达。表达产物采用常规层析方法纯化至约98%的纯度。两种商品化的酶和自行表达纯化T4DNA聚合酶均先用标准放射性同位素掺入法测定,国际单位(IU)定义为:1小时内使1nmol同位素标记的dATP掺入酸不溶性沉淀物所需的酶量定义为1个单位(U)。以放射性同位素标定后的T4DNA聚合酶作为标准品,稀释后按照实施例1的方法获得CT值并绘制标准曲线,如下表7。表7:不同来源的T4DNA聚合酶的CT值以及标准曲线的相关系数其中,商品化T4DNA聚合酶1曲线方程为:y=-0.876ln(x)+13.235,R2=0.9982(参见实施例1,曲线图请参阅图4)。商品化T4DNA聚合酶2曲线方程为:y=-0.881ln(x)+13.127,R2=0.9992,曲线图请参阅图5。自制T4DNA聚合酶曲线方程为:y=-0.877ln(x)+13.211,R2=0.9985,曲线图请参阅图6。上述结果表明,不同来源的T4DNA聚合酶的聚合酶均可形成标准曲线,且CT值彼此无明显差异。因此,这种标准曲线方法可作为聚合酶活性标定。参照实施例2的方法,检测未知T4DNA聚合酶活性的CT值,每组重复8个样品,结果如下表8。其中未知T4DNA聚合酶8组样品PCR扩增对应的CT值如表8所示。表8:未知T4DNA聚合酶样品的CT值未知样品标号1#2#3#4#5#6#7#8#CT值16.3116.2916.3316.3216.3416.2816.2716.3将未知T4DNA聚合酶对应的CT值分别带入商品化T4DNA聚合酶1曲线方程:y=-0.876ln(x)+13.235,商品化T4DNA聚合酶2曲线方程:y=-0.881ln(x)+13.127自制T4DNA聚合酶曲线方程:y=-0.877ln(x)+13.211。计算检样品中T4DNA聚合酶的酶活,结果如下表9。表9:未知样品中T4DNA聚合酶的酶活性上述结果表明,不同来源的T4DNA聚合酶均可采用上述检测方法进行检测,标准差以及变异系数小,表明检测的重现性好、灵敏度高。以上具体实施例以检测T4DNA聚合酶的结果为例说明,可以理解,上述检测方法对其他的DNA聚合酶也依然能够检测。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。SEQUENCELISTING110菲鹏生物股份有限公司、广东菲鹏生物有限公司120检测DNA聚合酶活性的方法及检测试剂盒1605170PatentInversion3.3210121193212DNA213人工序列4001aaatataataataagatcgaataatagatagagcagctgagctaccgtagccaaaggctg60ccgctgaacggatagagtacagcttgccacgat932102211117212DNA213人工序列4002cauauagcauaucgaucguagctgatcgtggcaagctgtactctatccgttcagcggcag60cctttggctacggtagctcagctgctctatctattattcgatcttattattatattt117210321124212DNA213人工序列4003catatagcatatcgatcgtagctg24210421123212DNA213人工序列4004aaatataataataagatcgaata23210521130212DNA213人工序列4005tggcaagctgtactctatccgttcagcggc30当前第1页1 2 3
免责声明 本文仅代表作者个人观点,与本网无关。其创作性以及文中陈述文字和内容未经本站证实,对本文以及其中全部或者部分内容、文字的真实性、完整性、及时性本站不做任何保证或承诺,请读者仅作参考,并请自行核实相关内容。
版权声明 未经蚂蚁淘授权不得转载、摘编或利用其他方式使用上述作品。已经经本网授权使用作品的,应该授权范围内使用,并注明“来源:蚂蚁淘”。违反上述声明者,本网将追究其相关法律责任。
发布于 : 2021-07-16
阅读(91)
▍
相关分类
▍
相关文章
质粒抽提经典中的经典从质粒抽提谈起
96孔板蒸发 96孔板振荡器_川汇区郑肖参电线五金电料批零店
求助,干细胞培养可以加抗生素吗? 神经干细胞专区干细胞之家 中国...
美国雅培Thermobrite原位杂交仪S500【价格,厂家,求购,使用说明...
核酸突变分析方法抉择:PCR、杂交、NGS?
什么是基因突变?
Domain Names Registered on 20091205_1 0912 2009 ...
碱性磷酸酶(AP)标记的羊抗牛IgG|Goat AntiBovine IgG/AP_上海科...
Shanghai Dongjin Plastic Fibre Products Co . Ltd ...
上海常海国际物流有限公司 国际海运网荣誉会员 ShippingChina
亚洲化学产业信息 | 化日(上海)投资咨询有限公司 | 亚洲 ...
领克特网络营销联盟 linktech.cn
▍
相关问答





